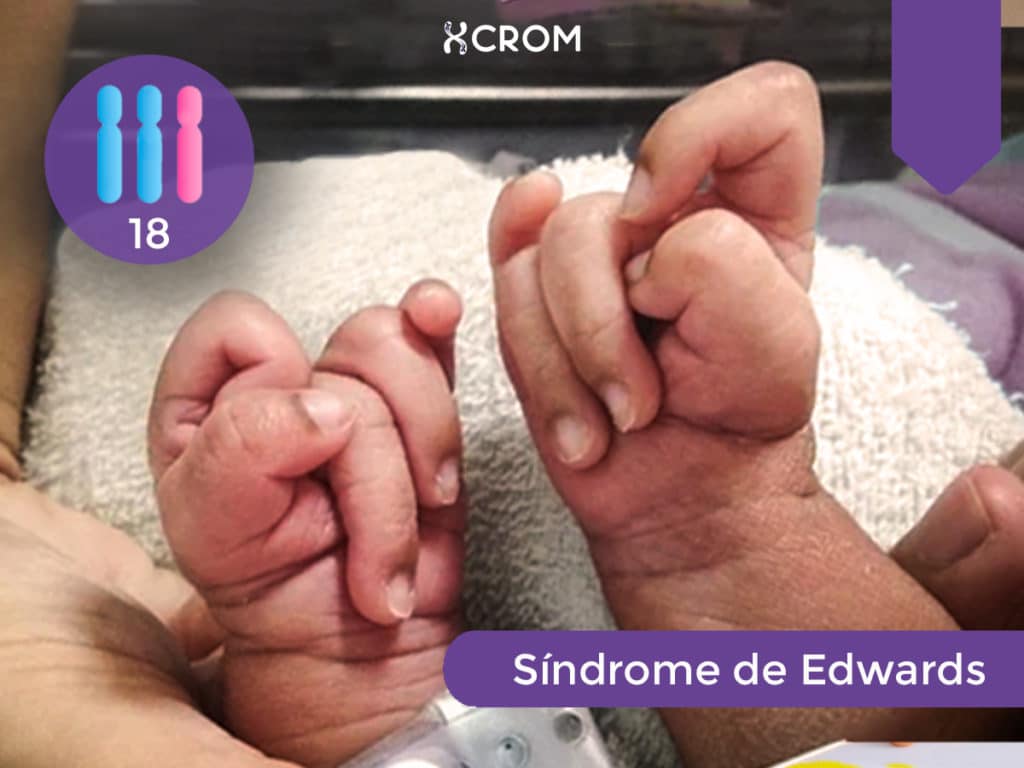
Por: Jennifer Pochne
El síndrome de Edwards, también conocido como trisomía 18, es un tipo de aneuploidía que fue originalmente descripta por John H. Edwards en un artículo publicado a comienzos de la década del 60 en la revista The Lancet perteneciente a la Universidad de Wisconsin.
Definición:
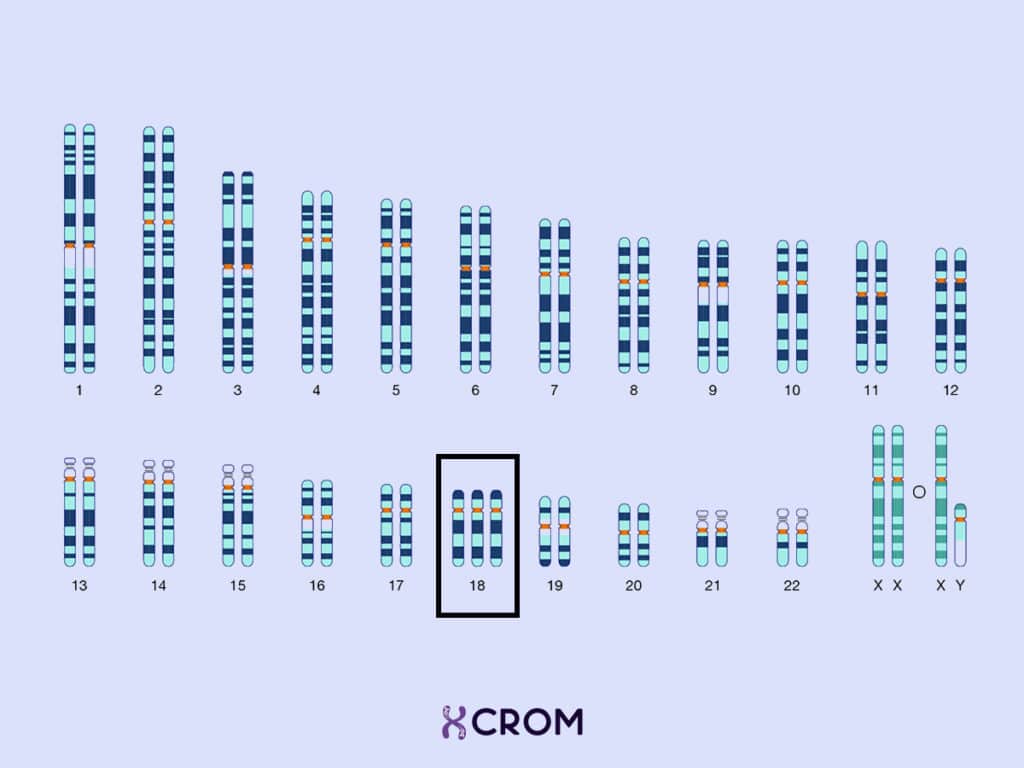
El síndrome de Edwards es un trastorno genético originado por un desbalance cromosómico representado por una trisomía 18. Las células de un individuo normal tienen 46 cromosomas: 23 cromosomas de origen materno y 23 cromosomas de origen paterno. Una persona que tiene síndrome de Edwards, tiene un cromosoma 18 adicional. Este material genético adicional es el causante de las características físicas y los problemas funcionales típicos del síndrome: retrasos serios en el desarrollo motor y cognitivo, discapacidad intelectual, tamaño pequeño al nacer y diversas malformaciones congénitas, como microcefalia grave, cardiopatías, occipital prominente, orejas de implantación baja y cara triangular característica.
Existen distintos tipos de síndrome de Edwards.
El 5% restante de los casos de este síndrome se distribuyen entre:
Prevalencia:
La incidencia del síndrome de Edwards es variable y se calcula entre 1:6.000 y 1:13.000 nacidos vivos, aunque los abortos espontáneos son frecuentes. Esta variabilidad está fuertemente condicionada por los contextos socioculturales. Por ejemplo, en países donde el aborto es ilegal la prevalencia es mayor, mientras que en aquellos donde el aborto es legal, la prevalencia baja. Por otra parte, la incidencia es similar en las diversas etnias y zonas geográficas, pero tal como sucede con otras trisomías, aumenta en función de la edad materna: a los 37 años, el riesgo de que nazca un bebé afecto del síndrome es de 1 de cada 2600 y este sigue en aumento a medida que avanza la edad de la mujer. Además, este síndrome es tres veces más frecuente en embriones de sexo femenino con respecto a embriones masculinos.
Fenotipo-clínica:
En los niños que presentan síndrome de Edwards el tamaño prenatal y al nacer es marcadamente pequeño para la edad gestacional, y tienen un llanto débil y una disminución de la respuesta al sonido. Los pacientes portadores de esta trisomía presentan algunas características físicas comunes:
Además de los rasgos faciales y físicos típicos, las personas con síndrome de Edwards suelen tener otros problemas. Entre los más comunes se encuentran:
Diagnóstico:
Existen dos tipos de pruebas prenatales que se utilizan para detectar el síndrome de Edwards: las pruebas de cribado y las pruebas diagnósticas.
Las pruebas de cribado calculan las probabilidades de que un feto tenga síndrome de Edwards. Estas son más económicas y fáciles de realizar, pero no permiten saber con certeza si un bebé tiene o no esta patología. Las pruebas de cribado suelen incluir una combinación de pruebas de sangre (donde se detectan sustancias específicas) y ecográficas (donde se mide por ejemplo la translucencia nucal) que, junto con la edad de la madre (las mujeres embarazadas de más de 35 años tienen más probabilidades de tener bebés con aneuploidías) y la edad gestacional del bebé, permite calcular el riesgo de que el bebé nazca con síndrome de Edwards. En la actualidad existe otra prueba de cribado que se basa en el análisis de ADN fetal circundante que se encuentra en la sangre de la madre. Esta última alternativa se puede hacer en el primer trimestre del embarazo y ofrece una precisión mucho más alta que los otros cribados.
Por otra parte, las pruebas diagnósticas permiten afirmar o negar con certeza si el feto padece esta anomalía y se basan en la realización de un cariotipo o análisis cromosómico: sobre una muestra biológica se realiza una observación de los cromosomas y se agrupan por tamaño, número y apariencia para buscar diferencias. Las pruebas diagnósticas durante el embarazo incluyen: el muestreo de vellosidades coriónicas (MVC), la amniocentesis, y el muestreo percutáneo de sangre umbilical o cordocentesis. Después del nacimiento, se puede realizar una prueba de cariotipo sobre una muestra de sangre del bebé.
Pronóstico:
Más del 90% de los recién nacidos diagnosticados con síndrome de Edwards mueren (la mitad de los bebés con esta afección no sobrevive más allá de la primera semana de vida), y el 95% no superan el primer año de vida. El 5% restante suele sobrevivir más tiempo: una vez que el niño cumple el año de edad, existe un 60% de probabilidad de que sobreviva más de 5 años. Las niñas presentan mayor tasa de supervivencia que los varones. Algunos pacientes han logrado sobrevivir hasta los años de la adolescencia, pero con problemas de salud y del desarrollo graves. Las complicaciones, que dependen de los defectos y síntomas específicos, pueden incluir: retraso mental y motor, dificultad para respirar o falta de respiración (apnea), sordera, problemas de alimentación y digestivos, insuficiencia cardíaca, escoliosis, convulsiones, problemas de la vista.
Tratamiento:
El síndrome de Edwards es una afección que no tiene un tratamiento específico, ya que los abordajes utilizados dependerán de la afección individual de la persona, y se debe tener en cuenta que solo un 5-10% sobrevive el primer año. Los pacientes que sobreviven tienen un marcado retraso del desarrollo y discapacidad, por lo que existe controversia sobre la realización de múltiples procedimientos invasivos para corregir diversas anomalías asociadas. Sí resulta fundamental la derivación temprana a fisioterapia y logoterapia, para mejorar la calidad de vida del paciente. Además, es imprescindible el apoyo y contención para la familia.