Por: Jennifer Pochne
El síndrome de Patau, también conocido como trisomía 13, es un tipo de aneuploidía que fue descubierta en 1960 por el genetista alemán Klaus Patau. Si bien esta trisomía se describió citogeneticamente en el siglo XX, las primeras observaciones sobre su existencia datan del siglo XVII: en 1656, el naturalista y fisiólogo Thomas Bartholin describió la clínica de un paciente que es compatible con las características de este síndrome.
Definición:
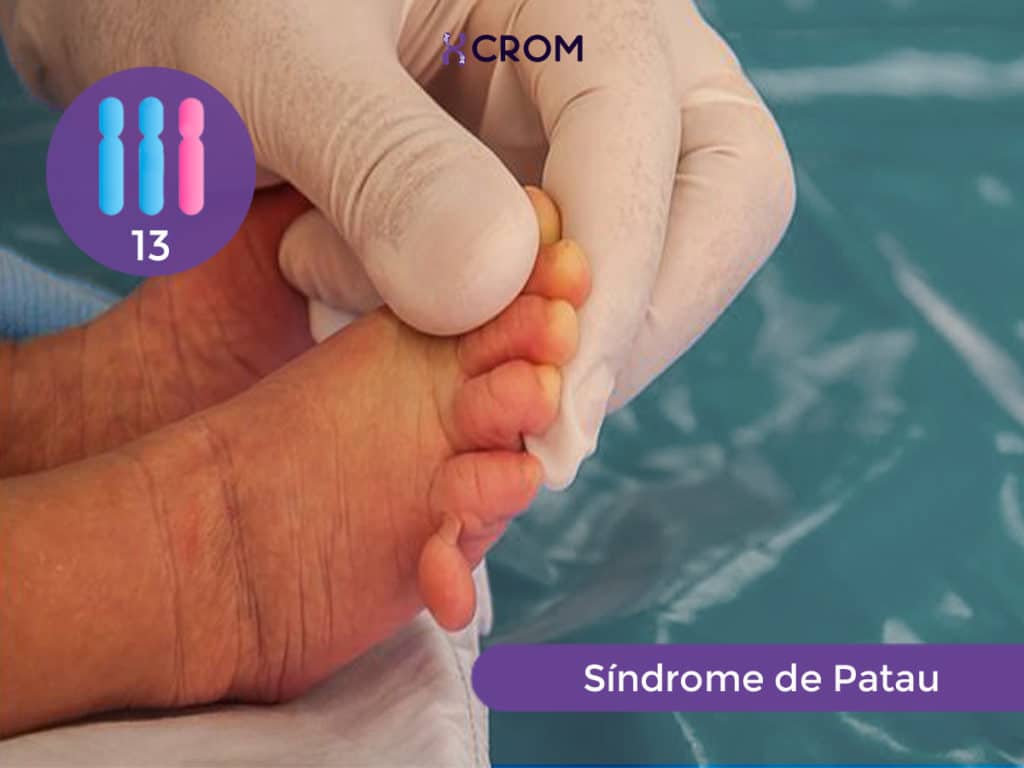
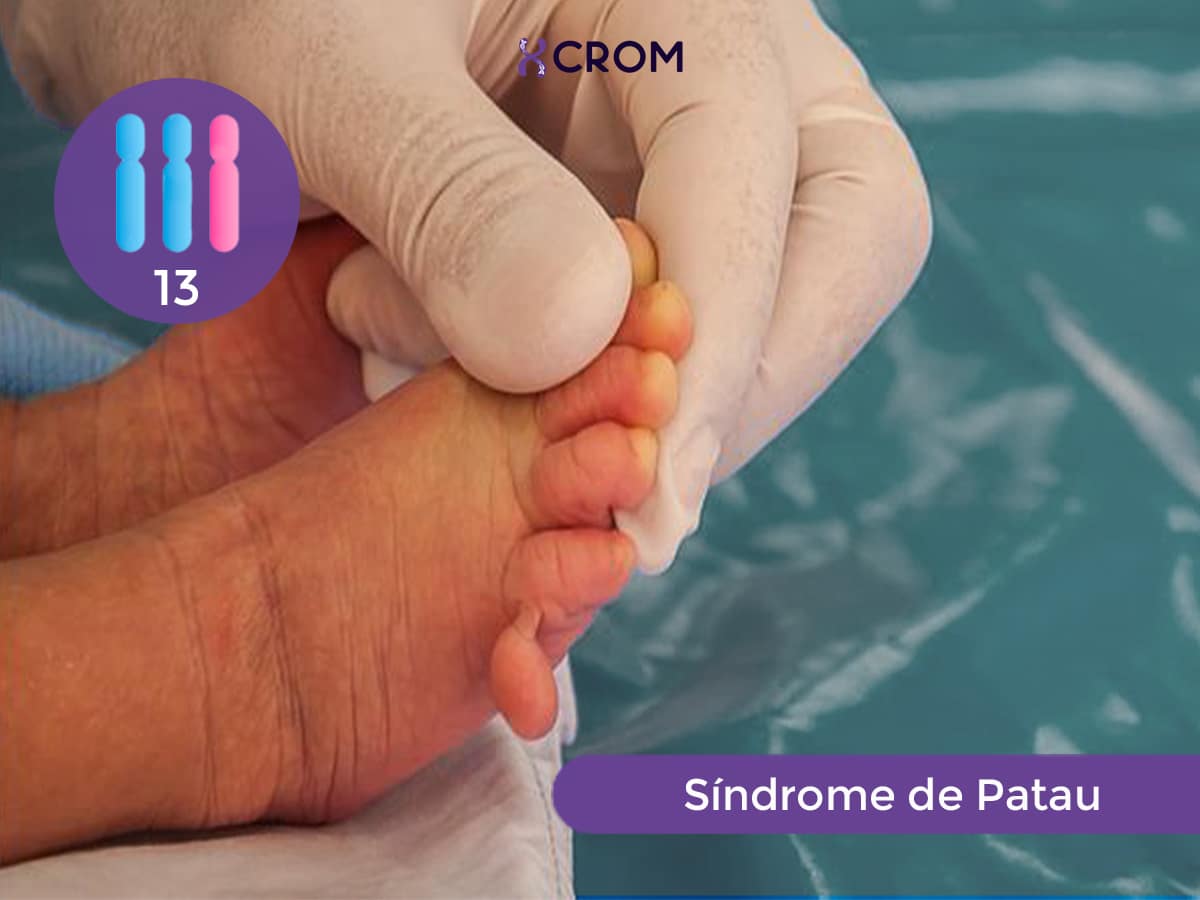
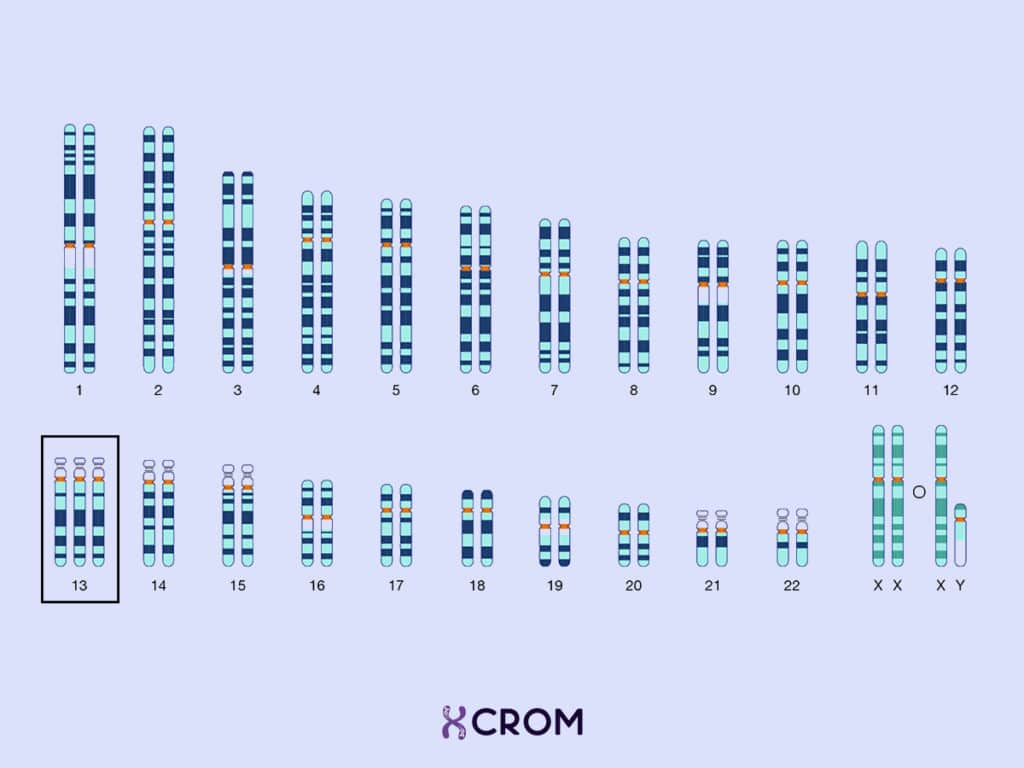
El síndrome de Patau es un trastorno genético originado por un desbalance cromosómico representado por una trisomía 13. Las células de un individuo normal tienen 46 cromosomas: 23 cromosomas de origen materno y 23 cromosomas de origen paterno. Una persona que tiene síndrome de Patau, tiene un cromosoma 13 adicional. Este material genético adicional es el causante de las características físicas y los problemas funcionales típicos del síndrome: tienen tamaño pequeño al nacer; poseen desarrollo anormal del prosencéfalo, la parte media de la cara y los ojos; manifiestan retraso en el desarrollo que implica al sistema nervioso (discapacidad intelectual grave), al musculoesquelético, al cardiovascular y al renal.
Tal como ocurre con otras trisomías, existen distintos tipos de síndrome de Edwards.
Prevalencia:
El síndrome de Patau es la trisomía reportada menos frecuente en la especie humana. Su incidencia se estima entre 1:8.000 y 1:15.000 nacimientos, aunque es más frecuente en abortos espontáneos y mortinatos: se cree que entre el 80-90% de los fetos con el síndrome no llegan a término. De hecho, la tasa de abortos espontáneos en fetos que presentan esta anomalía es tan elevada, que representa alrededor del 1% del total de abortos espontáneos reconocidos.
Los individuos que nacen con síndrome de Patau raramente superan el año de vida: la supervivencia oscila entre los 4 meses en los varones y los 20 meses en las mujeres. Alrededor del 50% de estos niños fallecen durante el primer mes de vida, y a los 6 meses han fallecido el 70% de los nacidos vivos.
Fenotipo-clínica:
Los recién nacidos con síndrome de Patau tienen retraso de crecimiento pre y post natal. Poseen un tamaño marcadamente pequeño al nacer y muestran un conjunto de malformaciones características que permiten la sospecha clínica en el momento del nacimiento. Los pacientes portadores de esta trisomía presentan algunas características físicas comunes, entre las que se encuentran:
Además de los rasgos faciales y físicos típicos, las personas con síndrome de Patau presentan otros tipos de malformaciones y problemas fisiológicos. Entre los más comunes se encuentran:

Diagnóstico:
Existen dos tipos de pruebas prenatales que se utilizan para detectar el síndrome de Patau: las pruebas de cribado y las pruebas diagnósticas.
Las pruebas de cribado calculan las probabilidades de que un feto tenga síndrome de Patau. Estas son más económicas y fáciles de realizar, pero no permiten saber con certeza si un bebé tiene o no esta patología. Las pruebas de cribado suelen incluir una combinación de pruebas de sangre (donde se detectan sustancias específicas) y ecográficas (donde se mide por ejemplo la translucencia nucal) que, junto con la edad de la madre (las mujeres embarazadas de más de 35 años tienen más probabilidades de tener bebés con aneuploidías) y la edad gestacional del bebé, permite calcular el riesgo de que el bebé nazca con síndrome de Patau. En la actualidad existe otra prueba de cribado que se basa en el análisis de ADN fetal circundante que se encuentra en la sangre de la madre. Esta última alternativa se puede hacer en el primer trimestre del embarazo y ofrece una precisión mucho más alta que los otros cribados.
Por otra parte, las pruebas diagnósticas permiten afirmar o negar con certeza si el feto padece esta anomalía y se basan en la realización de un cariotipo o análisis cromosómico: sobre una muestra biológica se realiza una observación de los cromosomas y se agrupan por tamaño, número y apariencia para buscar diferencias. Las pruebas diagnósticas durante el embarazo incluyen: el muestreo de vellosidades coriónicas (MVC), la amniocentesis, y el muestreo percutáneo de sangre umbilical o cordocentesis. Después del nacimiento, se puede realizar una prueba de cariotipo sobre una muestra de sangre del bebé.
Pronóstico:
Los pacientes con Síndrome de Patau tienen una supervivencia muy escasa: aproximadamente el 28% muere en la primera semana de vida, el 44% en el primer mes y el 86-90% en el primer año. Sólo el 5% sobrevive más de 3 años, y se sabe que el pronóstico de vida de los pacientes se relaciona claramente con la gravedad de las malformaciones cerebrales, renales y cardiacas.
Se ha registrado supervivencia prolongada (en algunos casos hasta la edad adulta), pero esta situación es más común en casos de trisomía en mosaico o parcial y en ausencia de malformaciones graves del cerebro. En general, los pacientes sin mosaicismo solo desarrollan autonomía limitada, con ausencia de habla y deambulación.
Tratamiento:
Los pacientes que nacen con síndrome de Patau y sobreviven necesitan asistencia médica desde el mismo momento del nacimiento. Si bien las anomalías cardiacas representan la causa principal de morbimortalidad en estos pacientes, se plantea el problema ético de si su reparación quirúrgica está indicada, teniendo en cuenta el pésimo pronóstico del cuadro tanto desde el punto de vista físico como intelectual.
Menos del 10% de los nacidos sobrevive el año de vida y es dado de alta. Estos pacientes precisan de atención especializada en el domicilio, requiriendo la intervención de un equipo multidisciplinar. En estos casos resulta imprescindible el apoyo, la contención y la capacitación de los padres y la familia, que han de ser previamente entrenados para la realización de determinadas tareas y maniobras que pueden ser de importancia vital para la supervivencia del paciente. El tratamiento solo es de apoyo, ya que no existen posibilidades de revertir las condiciones.